L’atrofia muscolare spinale, conosciuta anche come SMA, è una malattia genetica rara che colpisce il sistema nervoso, causando una perdita graduale dei motoneuroni. Di natura ereditaria, la SMA è causata da mutazioni nei geni SMN1 o SMN2, che hanno il compito di produrre una proteina essenziale per la sopravvivenza dei motoneuroni. Ci sono cinque forme distinte di atrofia muscolare spinale: tipo 0, tipo 1, tipo 2, tipo 3 e tipo 4. Le prime tre sono gravi, e spesso portano al decesso precoce, mentre le varianti tipo 3 e tipo 4 sono meno gravi, infatti influenzano la qualità della vita senza causare la morte anticipata. Ma vediamo tutte le caratteristiche di questa malattia e i vari trattamenti disponibili.
Cos’è, come avviene la diagnosi e i trattamenti per l’atrofia muscolare spinale
Conosciuta anche come SMA, l’atrofia muscolare spinale è una rara malattia genetica neurodegenerativa che si caratterizza per la perdita progressiva dei motoneuroni spinali e del tronco encefalico. Questa patologia si presenta con atrofia e conseguente indebolimento dei muscoli scheletrici, portando a difficoltà motorie.
La SMA può risultare fatale in giovane o giovanissima età, soprattutto nelle sue forme più gravi. Queste varianti della malattia compromettono l’efficienza dei muscoli respiratori, causando episodi di insufficienza respiratoria o polmonite, con esito spesso mortale.
I neuroni motori, noti anche come motoneuroni, sono cellule nervose originarie del sistema nervoso centrale, comprendente l’encefalo e il midollo spinale. Attraverso i loro prolungamenti, gli assoni, regolano l’attività di muscoli e ghiandole.
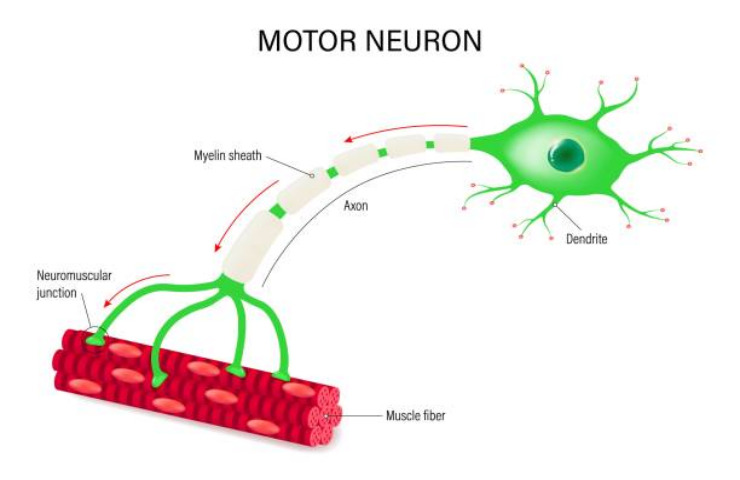
Esistono due categorie di motoneuroni: quelli superiori (o primi motoneuroni) e quelli inferiori (o secondi motoneuroni). I motoneuroni superiori hanno origine nell’encefalo e controllano l’attività dei motoneuroni inferiori, che principalmente derivano dal midollo spinale e gestiscono l’attività dei muscoli scheletrici, dei muscoli lisci, del muscolo cardiaco e del cuore.
Nei soggetti affetti da SMA, i motoneuroni subiscono una degenerazione progressiva, causando un’atrofia muscolare da mancato utilizzo che, nelle forme più gravi, può manifestarsi come paralisi, insufficienza respiratoria e, in ultima istanza, decesso.
L’atrofia muscolare spinale si sviluppa a causa di mutazioni nel gene SMN1, che si verifica nella maggioranza dei casi, o nel gene SMN2, più raramente. Entrambi questi geni sono localizzati sul cromosoma 5 e sono responsabili della sintesi della proteina nota come SMN, fondamentale per la sopravvivenza dei motoneuroni. Come indica il nome della proteina generata da SMN1 e SMN2, le mutazioni in questi geni privano i motoneuroni di una sostanza biologica essenziale per la loro sopravvivenza, riducendo i livelli proteici. Ad esempio, in presenza di mutazioni in SMN1, i livelli di proteina SMN diminuiscono al 10-20% rispetto alla norma.
Naturalmente, la carenza di quantità adeguate della proteina SMN porta alla progressiva degenerazione dei motoneuroni. La perdita di questi neuroni interrompe la trasmissione nervosa che regola l’attività muscolare nel corpo umano. Di conseguenza, i muscoli, in quanto non più funzionali, subiscono un processo graduale di atrofia e indebolimento.
Ma vediamo nel dettaglio i tipi di atrofia muscolare spinale. In base all’età in cui si manifesta e alla gravità della condizione, gli specialisti identificano cinque diverse varianti di atrofia muscolare spinale:
- La SMA di tipo 0: è la forma più estrema, manifestandosi prima della nascita con limitata mobilità del feto. Solitamente, i neonati affetti sopravvivono solo poche settimane, anche con l’ausilio di supporto respiratorio. Al momento della nascita, il neonato affetto mostra chiare difficoltà nel processo di deglutizione e nella respirazione.
- La SMA di tipo 1: è la variante più grave e comune che si presenta durante la vita (circa il 50% dei casi). Si manifesta tipicamente entro il sesto mese di vita, con un’alta probabilità di causare il decesso nei primi anni o, in rari casi, durante l’adolescenza. La morte avviene generalmente a causa di insufficienza respiratoria o infezioni polmonari. I piccoli con SMA di tipo 1 manifestano una muscolatura estremamente fragile e che non si sviluppa in modo normale (atrofia muscolare). Questo compromette la capacità di eseguire azioni come sollevare il capo, muovere gli arti e mantenere una posizione seduta. Inoltre, progressivamente complica funzioni vitali come succhiare il latte, deglutire, masticare e respirare.
- La SMA di tipo 2: rappresenta la seconda forma per gravità. Di solito, compare tra i 7 e i 18 mesi di vita. Rispetto alla SMA di tipo 1, la prospettiva di vita è più lunga, consentendo ai pazienti di raggiungere l’età adulta. La SMA di tipo 2 si presenta tipicamente con:
– Debolezza muscolare negli arti superiori e inferiori;
– Tremori alle dita e alle mani;
– Complessità nell’assumere autonomamente la posizione seduta (sebbene il paziente riesca a mantenerla);
– Ostacoli nel sollevarsi in piedi e camminare;
– Deformità e problematiche articolari;
– Sfide nella respirazione e nella deglutizione del cibo;
– Insorgenza di scoliosi (solitamente, più tardi nella progressione della malattia).
In questa situazione, le difficoltà respiratorie e di deglutizione del cibo costituiscono la principale causa di morte prematura, che solitamente si verifica all’inizio dell’età adulta.
- La SMA di tipo 3: meno grave delle due precedenti, questa variante si manifesta di solito dopo i 18 mesi di vita, ma in alcuni casi può emergere anche durante l’infanzia o l’adolescenza. Sebbene comporti disabilità significative, non incide sull’aspettativa di vita. La variante di SMA di tipo 3 genera problemi nella postura e nell’equilibrio, tremolii alle mani e difficoltà nell’ergersi da una posizione seduta, nel camminare, nel salire le scale e nel correre.
- La SMA di tipo 4: costituisce la forma adulta e meno grave della malattia. Solitamente, inizia intorno alla terza decade di vita con un andamento molto lento. Non solitamente associata a problemi respiratori, presenta un’aspettativa di vita normale. Inizia in età adulta, la SMA di tipo 4 è comunemente caratterizzata da:
– Diminuzione del tono muscolare, coinvolgendo braccia e gambe;
– Ostacoli nel camminare;
– Tremolii e contrazioni muscolari improvvisi.
La gravità della SMA è influenzata dai livelli di proteina SMN, con una maggiore riduzione della quantità di SMN correlata a una maggiore severità della malattia. La diminuzione dei livelli di SMN è strettamente legata all’entità del difetto genetico che ha colpito i geni SMN1 o SMN2, con una delezione genica, ad esempio, che può causare una significativa riduzione della proteina SMN.
Come viene effettuata la diagnosi? Per confermare la presenza di atrofia muscolare spinale, è essenziale eseguire un test genetico utilizzando un campione di sangue del paziente.
L’esecuzione del test genetico per identificare la SMA è generalmente giustificata da un esame obiettivo e da una storia medica che suscitano sospetti, evidenziando sintomi e una storia familiare associati all’atrofia muscolare spinale.
Occasionalmente, durante la diagnosi di SMA, possono essere impiegati altri test come l’elettromiografia o la biopsia muscolare.
Esiste una cura per la SMA? La gestione terapeutica della SMA è un argomento complesso. Fino a pochi anni fa, la malattia risultava incurabile. Tuttavia, al giorno d’oggi, è disponibile una cura fondata sulla terapia genica, che mira a correggere la mutazione responsabile della SMA e a preservare i motoneuroni dalla loro graduale degenerazione.
Tuttavia, questa cura rivoluzionaria presenta due limitazioni cruciali, legate ai costi e all’applicabilità, che continuano a complicare l’approccio terapeutico alla SMA e ad ostacolare una possibilità di guarigione estesa a tutti i pazienti.
Considerando queste sfide, l’approccio terapeutico ancora ampiamente adottato tra i pazienti con atrofia muscolare spinale rimane quello sintomatico. Questo si basa su trattamenti finalizzati ad alleviare la sintomatologia e le complicanze, con l’obiettivo generale di migliorare la qualità di vita dei malati.
